






Abstract
The histopathologic features of 10 myxoid adrenocortical neoplasms were analyzed, and epidermal growth factor receptor (EGFR) expression, EGFR gene copy number, and EGFR gene mutations in the 10 tumors were detected by using immunohistochemical analysis, fluorescence in situ hybridization, and the Scorpion Amplification Refractory Mutation System (DxS, Manchester, England), respectively. Histologically, all 10 tumors varied in their myxoid composition, ranging from 20% to 95%. EGFR protein overexpression was more frequent in myxoid adrenocortical carcinomas (3/4) than in myxoid adrenocortical adenomas (0/6). However, EGFR mutations and EGFR amplification were rare. All patients with adenomas survived for the follow-up period with no recurrence of their tumors or evidence of metastatic disease; 3 patients with carcinoma died of the disease, and 1 is alive with disease. Myxoid changes in adrenocortical neoplasms can be present in adenomas and carcinomas. Detection of EGFR protein expression may be useful in the differential diagnosis between adrenocortical adenomas and adrenocortical carcinomas.
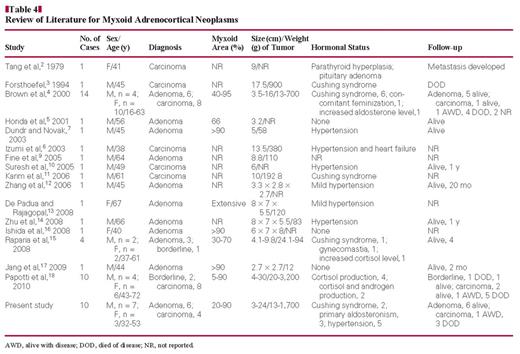
Adrenocortical adenomas (ACAs) and adrenocortical carcinomas (ACCs) show a variety of histopathologic patterns ranging from diffuse to alveolar, with a trabecular pattern the most common. They are sometimes admixed in the same neoplasm.1 A rare histologic variant is the myxoid type. Tang et al2 first reported a case of myxoid ACC in 1979, and several cases have been reported during the following 30 years. To date, including adenomas and carcinomas, a total of 41 cases (21 carcinomas, 3 borderline malignancies, and 17 adenomas) have been reported in the English literature.2–18 Because the presence of myxoid changes in adrenocortical neoplasms often raises the possibility of malignancy, we aimed to explore the clinicopathologic and molecular biologic features of myxoid adrenocortical neoplasms in this study.
The epidermal growth factor receptor (EGFR) pathway has been proposed to be important for cancer pathophysiology. EGFR belongs to the ErbB family of tyrosine kinase receptors, which have strong regulatory effects on cell proliferation, differentiation, survival, and migration.19 In ACCs, EGFR overexpression and EGFR point mutations have also been described.20,21 However, there has been no analysis of EGFR protein expression, somatic mutations, and amplification in myxoid adrenocortical neoplasms.
In this study, we analyzed the clinicopathologic and immunohistochemical features of 10 myxoid adrenocortical neoplasms (6 benign and 4 malignant). Furthermore, we explored EGFR protein expression, EGFR mutations, and EGFR copy number in these rare tumors. We also reviewed the literature detailing these unusual neoplasms.
Materials and Methods
Case Selection
The records of 989 consecutive patients seen at the Peking Union Medical College Hospital, Beijing, People’s Republic of China, from 2000 to 2009 with adrenocortical tumors were reviewed. The myxoid adrenocortical neoplasms were defined according to previous study4 and our examination, and 10 cases of tumors with 10% or more myxoid areas were included in the study. For EGFR analysis, 5 cases of conventional ACA and 5 cases of conventional ACC were used as control cases. Follow-up data were available for all patients. The Peking Union Medical College Hospital Ethics Committee approved this study.
Histochemical and Immunohistochemical Studies
Paraffin-embedded tissue sections were stained with alcian blue (pH, 2.5), periodic acid–Schiff (PAS), and mucicarmine stains.
EGFR protein expression was evaluated by using immunohistochemical studies with a mouse antihuman EGFR monoclonal antibody (clone 2-18C9; Pharm Dx Kit; DAKO, Carpinteria, CA) according to the manufacturer’s instructions. Sections (4 μm thick) were cut from paraffin tissue blocks, deparaffinized in xylene, and rehydrated in decreasing concentrations of ethanol. Appropriate positive and negative control samples were used. Positive results were defined as greater than 10% tumor cells showing membranous staining of any intensity. The percentage of positive cells and intensity, defined as mild (1+), moderate (2+), or strong (3+), was recorded for each case.
Immunohistochemical evaluation of the expression of pan-cytokeratin (cloneAE1/AE3, prediluted; Novocastra, Newcastle upon Tyne, England), vimentin (clone V9, dilution 1:500; DAKO, Glostrup, Denmark), α-inhibin (clone ZM-0460, prediluted; Santa Cruz Biotechnology, Santa Cruz, CA), Melan-A (clone A103, dilution 1:50; DAKO, Glostrup, Denmark), synaptophysin (clone ZA-0236, prediluted; Invitrogen, San Diego, CA), chromogranin A (clone 5H7, prediluted, Novocastra), and Ki-67 (clone SP6, prediluted; Santa Cruz Biotechnology) was performed using a Ventana Benchmark XT instrument (Ventana, Tucson, AZ). The tissue sections were mounted on charged glass slides, and the instrument automated the immunostaining, including baking, dewaxing, rehydration, blocking of endogenous peroxidase, incubation in primary and secondary antibodies, label and color developer, and washings with tris(hydroxymethyl)aminomethane buffer. All other reagents were obtained from Ventana. Appropriate positive and negative control samples were used.
Immunohistochemical reactivity for Ki-67 was recorded as the percentage of stained nuclei. The positivity for cytokeratin (pan), vimentin, α-inhibin, Melan-A, chromogranin A, and synaptophysin was defined as cytoplasmic or membranous staining.
DNA Extraction and EGFR Mutations Using the Scorpion Amplified Refractory Mutation System
Formalin-fixed, paraffin-embedded tumor sections were deparaffinized and air dried. Tumor cells were acquired by microdissection from 6 to 8 slides from each myxoid adrenocortical neoplasm. Genomic DNA was extracted using standard Proteinase K digestion and a DNeasy minispin column (TIANamp Genomic DNA Kit, Tiangen Biotech, Beijing, China).
We detected 29 specific EGFR mutations in exons 18 through 21 by using the EGFR Mutation Test Kit (DxS, Manchester, England) and the Scorpion Amplified Refractory Mutation System (DxS). The assays that used allele-specific real-time polymerase chain reaction were carried out according to the manufacturer’s protocol using an Applied Biosystems (Foster City, CA) 7500 real-time polymerase chain reaction system.22–24 Data analysis was performed using Applied Biosystems SDS software.
Fluorescence In Situ Hybridization
EGFR fluorescence in situ hybridization (FISH) analysis was carried out using the LSI EGFR SpectrumOrange/CEP 7 SpectrumGreen probe (Vysis, Abbott Laboratories, Downers Grove, IL) according to the manufacturer’s protocol. Sections were incubated at 56°C overnight, deparaffinized by washing in CitriSolv (Fisher Scientific, Pittsburgh, PA), and dehydrated in 100% ethanol. After incubation in 2× saline sodium citrate buffer (2× SSC, pH 7.0) at 75°C for 15 to 25 minutes, the sections were digested with Proteinase K (0.25 mg/mL in 2× SSC; pH 7.0) at 37°C for 15 to 25 minutes, rinsed in 2× SSC at room temperature for 5 minutes, and dehydrated in a series of increasing concentrations of ethanol (70%, 85%, and 100%). The EGFR/CEP 7 probe set was applied to areas on each slide selected on the basis of tumor foci, and the hybridization area was covered with a glass coverslip and sealed with nail polish. The slides were incubated at 80°C for 8 to 10 minutes for codenaturation of the chromosomal and probe DNA and then hybridized at 37°C for 20 to 24 hours.
Posthybridization washes were performed in 1.5 mol/L urea and 0.1× SSC (pH 7.0–7.5) at 45°C for 30 minutes and in 2× SSC for 2 minutes at room temperature. After the samples were dehydrated in ethanol, 4′,6′-diamidino-2-phenylindole (DAPI) in phosphate-buffered saline and glycerol (Vysis) was applied for chromatin counterstaining. FISH analyses were performed independently by 2 of us (J.G. and X.Z.) who were blinded to the clinical characteristics of the patients and to all other molecular variables. For EGFR FISH analyses, 60 nuclei were scored for signals from both DNA probes using an Olympus BX51TRF microscope (Olympus, Tokyo, Japan) equipped with a triple-pass filter (DAPI/green/orange, Vysis) at a final magnification of ×1,000.
EGFR gene status was classified into 6 categories based on the frequency of tumor cells with specific copy numbers of the EGFR gene and chromosome 7 centromere, as described previously25: (1) disomy (<2 copies in >90% of cells), (2) low trisomy (<2 copies in >40% of cells, 3 copies in 10%–40% of cells, and >4 copies in <10% of cells), (3) high trisomy (<2 copies in >40% of cells, 3 copies in >40% of cells, and >4 copies in <10% of cells), (4) low polysomy (>4 copies in 10%–40% of cells), (5) high polysomy (>4 copies in >40% of cells), and (6) gene amplification (presence of tight EGFR gene clusters and a ratio of the EGFR locus to chromosome 7 >2.0 or >15 copies of EGFR per cell in at least 10% of cells). Based on EGFR gene status, cases were further classified into 2 groups: (1) EGFR FISH–negative or low gene copy (disomy, low trisomy, high trisomy, and low polysomy) or (2) EGFR FISH–positive or high gene copy (high polysomy and gene amplification). For each FISH preparation, known positive and negative cells were used as controls.
Results
The ages of patients ranged from 32 to 53 years, with mean age of 41.9 years. The overall male/female ratio was 2.3:1 (2:1 for ACAs and 3:1 for ACCs). All patients had hormonal abnormalities, 2 patients had Cushing syndrome (2 with an ACC), 3 had primary hyperaldosteronism (2 with an ACA and 1 with an ACC), and 5 had hypertension (4 with an ACA and 1 with an ACC). The follow-up period ranged from 2 to 38 months, with a mean of 19.3 months for all patients. All 6 patients with ACA remained alive during the follow-up period, and none had any local recurrence or metastatic disease. Of the patients with ACC, 3 died as a result of the disease, and 1 was alive with disease at last follow-up. The clinical findings for the 10 cases of myxoid adrenocortical neoplasms are summarized in Table 1.
Macroscopic Findings
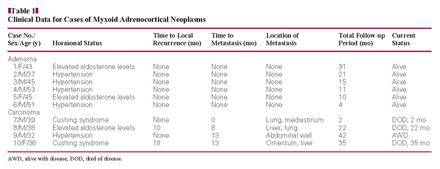
Grossly, the ACAs ranged in size from 3.0 to 6.5 cm and weighed from 11 to 52 g. In contrast to ACAs, the ACCs ranged in size from 7.0 to 24 cm and weighed from 45 to 2,480 g. It was obvious that the ACAs were generally smaller than the ACCs, weighing less than 100 g and measuring 6.5 cm or less in greatest diameter. The 6 ACAs were well encapsulated, and the 4 ACCs were partially encapsulated. On the cut surface, the majority of ACAs appeared yellow-grayish with gelatinous myxoid areas admixed with firm and fibrous areas Image 1A; areas of hemorrhage and necrosis were not observed. In contrast, the ACCs showed obvious areas of hemorrhage and necrosis Image 1B.
Histologically, the neoplasms were characterized by a myxoid background containing a distinct pattern of tumor cells that were floating loosely in an acellular myxoid background Image 1C, Image 1D, Image 1E, and Image 1F. All 10 tumors had prominent myxoid matrix material ranging from 20% to 90%. The tumor cells were mostly arranged in thin, delicate, anastomosing cords or floating clusters; some formed a pseudoglandular architecture. Other tumors had a nested pattern, with the constituent cells separated from their fibrous stromal frameworks by a variable amount of myxoid matrix. The cells appeared to float loosely in a copious acellular myxoid background divided into small lobules by thin, fibrovascular stroma. The tumor cells were polygonal with distinct cell borders and a moderate amount of clear, finely granular eosinophilic or amphophilic cytoplasm. The nuclei were enlarged, round to oval, hyperchromatic, and showed mild (in the adenomas) or moderate pleomorphism with nuclear variability in some areas (in the carcinomas). In all cases, the cells retained ample cytoplasm that was predominantly eosinophilic, with occasional clear cells. Necrosis and vascular invasion were absent in adenomas. Although 1 tumor had focal areas of capsular invasion, 4 ACCs had areas of confluent necrosis, obvious areas of hemorrhage, and capsular invasion Image 1G, and 3 tumors showed vascular invasion. Mitoses numbered 2 or fewer per 10 high-power fields (HPFs) among the ACAs, whereas the ACCs had 4 or more mitoses per 10 HPFs Image 1H. The pathologic findings of the ten cases are summarized in Table 2.
Histochemical and Immunohistochemical Findings
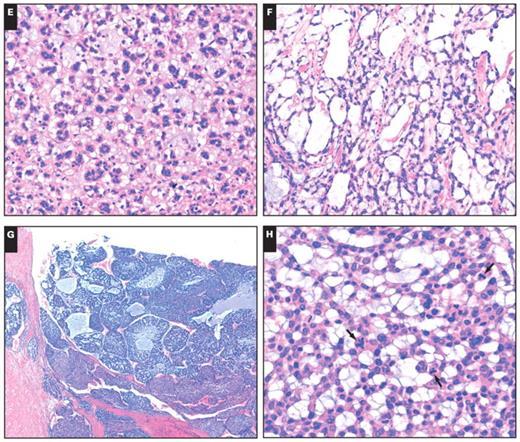
Alcian blue staining showed the presence of extracellular acidic mucosubstances in the acellular myxoid areas in all 10 cases Image 2A. Focal staining with PAS was present in 1 tumor, and focal staining for mucicarmine was observed for 1 ACA and 1 ACC. None of the histochemical stains demonstrated intracellular mucosubstances, suggesting that the myxoid material is a stromal product.
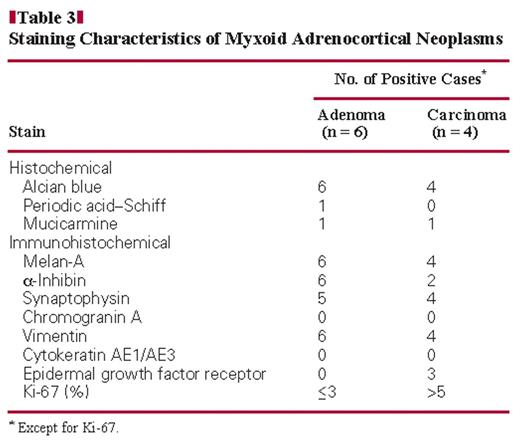
All 10 tumors stained for Melan-A Image 2B and vimentin. Staining for synaptophysin was positive in all cases except 1 ACA. Of the 10 tumors, 8 were focally to diffusely positive for α-inhibin Image 2C. All tumors were negative for cytokeratins and chromogranin A. Membranous EGFR was positive in 3 of 4 ACC cases Image 2D, whereas no ACAs were positive for EGFR. There was a significant difference in EGFR expression between the ACAs and ACCs, and EGFR overexpression mainly occurred in ACCs, which were consistent with conventional tumors (3 of 5 ACCs showed EGFR overexpression; 2 ACAs showed weak EGFR expression). The Ki-67 proliferative index ranged from 6% to 30% with a mean of 17.8% in all ACCs. However, all ACAs showed a low proliferative index (no more than 3%). Table 3 summarizes the results of the histochemical and immunohistochemical staining of paraffin-embedded sections from the 10 tumors.
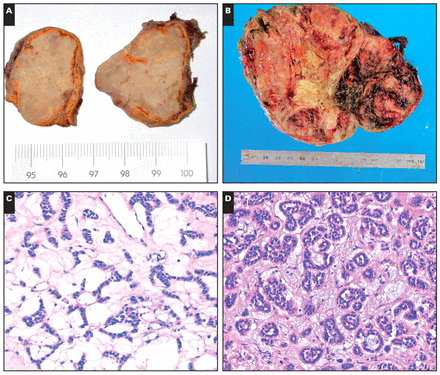
A, Gross photograph of a myxoid adrenocortical adenoma. On the cut surface, the majority of the adrenocortical adenoma had yellow-grayish regions with gelatinous myxoid areas admixed with areas of firm and fibrous consistency. B, Gross photograph of a myxoid adrenocortical carcinoma. The section showed obvious areas of hemorrhage and necrosis. C, A myxoid adrenocortical adenoma showed anastomosing cords of tumor cells (H&E, ×200). D, Prominent pseudoglandular pattern in a background of myxoid stroma (H&E, ×200). E, In the myxoid matrix are floating clusters of cells (H&E, ×200). F, A microcystic pattern in the myxoid background (H&E, ×100). G, A myxoid adrenocortical carcinoma demonstrated sheets and solid patterns and obvious areas of hemorrhage (H&E, ×10). H, Under high magnification, the neoplastic cells showed abnormal mitosis (arrows) (H&E, ×400).
EGFR Mutations and Copy Number
None of the 29 specific EGFR mutations in exons 18 through 21 that were tested for were detected in the ACAs and ACCs. All cases showed wild-type curves Image 3.
None of the 10 cases demonstrated EGFR amplification, and only 1 ACA showed high polysomy Image 4 (>4 copies in >40% of the cells). Two cases of myxoid tumors (including 1 ACA and 1 ACC) showed low polysomy, and 7 cases showed disomy, consistent with conventional tumors (2 of 5 ACCs showed high polysomy; all 5 ACAs showed disomy). The standard for FISH positivity was based on the criteria of Cappuzzo et al.25
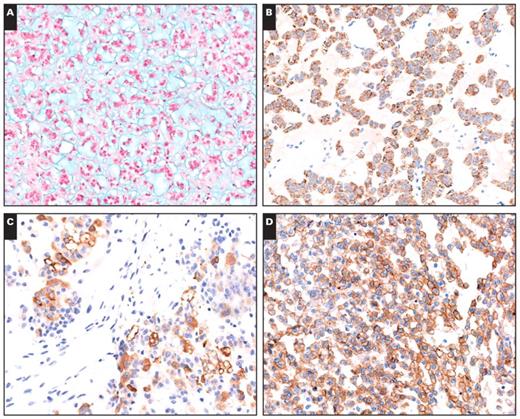
A, Alcian blue staining showed extracellular acidic mucosubstances in the acellular myxoid areas in all cases (×200). B, Immunohistochemical positivity for Melan-A staining in a myxoid adrenocortical adenoma. The myxoid areas between the cortical cells are negative (×200). C, A myxoid adrenocortical carcinoma was positive for α-inhibin (×200). D, Immunohistochemical positivity for epidermal growth factor receptor in the tumor cells of a myxoid adrenocortical carcinoma (×200).
Discussion
Myxoid ACC was first reported by Tang et al2 in 1979, and a second case was described 15 years later by Forsthoefel3 in 1994. In 2000, Brown et al4 described 14 cases of myxoid adrenocortical neoplasms, including 6 adenomas and 8 carcinomas, which was the largest study reported to date. Because these tumors are rare, the majority of the literature consists of case reports detailing ACAs and ACCs Table 4. To date, there have been no studies examining the molecular biologic features of myxoid adrenocortical neoplasms. In this study, we examined EGFR protein expression, EGFR mutations, and EGFR copy number in myxoid ACCs and ACAs. Our study of 10 myxoid adrenocortical neoplasms demonstrates that their clinicopathologic features are uncommon and that EGFR protein was overexpressed specifically in myxoid ACCs.
The myxoid adrenocortical neoplasms reported to date (51 cases, including our 10 cases; Table 4) occurred in patients ranging in age from 16 to 72 years, with a mean age of 48.7 years. The male/female ratio was 1.25:1, with 27 males and 24 females. Most of the reported cases of myxoid adrenocortical neoplasms (36/51) showed increased serum hormone levels, including cortisol, aldosterone, adrenocorticotropic hormone, follicle-stimulating hormone, and/or luteinizing hormone. In the present study, 2 patients had Cushing syndrome, 3 had primary hyperaldosteronism, and 5 had hypertension. Of the 51 cases reported to date, there were 23 adenomas, 3 borderline neoplasms, and 25 carcinomas. Typical and myxoid ACCs had a poor prognosis: Of the 25 patients with ACC, 13 died during the follow-up period, 4 were alive with metastatic disease, 4 were alive with no metastases, and for 4, there were no follow-up data. Early metastasis often occurs, especially to the lung and liver. In contrast, myxoid ACAs showed a good prognosis, with an almost 100% survival rate. Of the 23 cases reported to date, 19 patients survived the follow-up period; there were no follow-up data for 3. There were 3 patients diagnosed with borderline myxoid adrenocortical neoplasms of uncertain malignant potential in 2 studies.15,18 Of these 3 patients, 2 survived the follow-up period and 1 died.
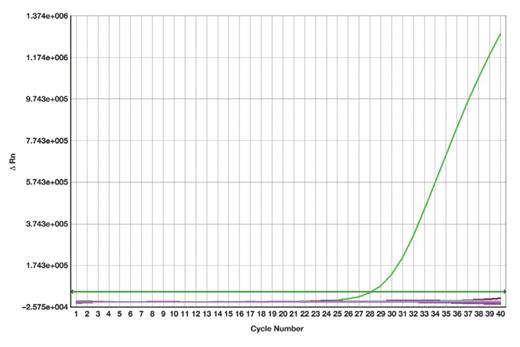
(Case 5) No mutation was found in a myxoid adrenocortical neoplasm. One ascending curve represented a wild-type of epidermal growth factor receptor detected by Scorpion Amplified Refractory Mutation System.
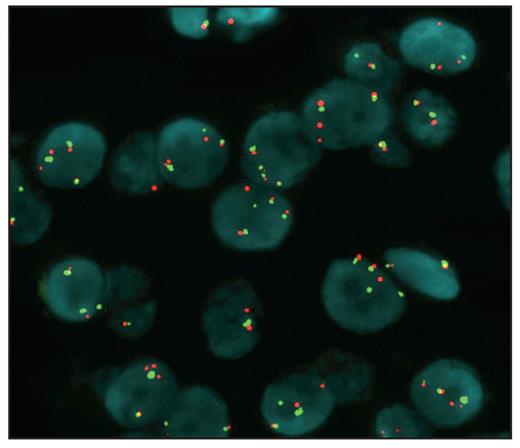
The myxoid adenoma cells show high polysomy of epidermal growth factor receptor (EGFR) by fluorescence in situ hybridization. Green signals represent the chromosome 7 centromere; red signals, the EGFR gene.
Grossly, as conventional adrenocortical tumors, adenomas are generally smaller than carcinomas, weighing less than 100 g and measuring 7.5 cm or less in the greatest diameter. Histologically, the usual adrenocortical neoplasms have architectural patterns ranging from diffuse to alveolar, with a trabecular pattern the most common. The rare myxoid variant is composed of polygonal or elongated cells with moderate amounts of eosinophilic cytoplasm. The cells usually are arranged in delicate arborizing cords or trabeculae, 1 to 2 cells thick surrounded by relatively large, acellular, clear myxoid spaces resembling the background of a myxoma.
The histochemical profile of the myxoid material in our series was consistent with the profile reported in previous case reports,2–8 with prominent alcian blue positivity of the extracellular mucosubstances and negative or focal weak staining with PAS and mucicarmine. With regard to the immunophenotypic profile, the cases in this study and those reported in the literature demonstrated vimentin, Melan-A, synaptophysin, and α-inhibin overexpression.15 In our cases and the cases reported in the literature, these tumors did not demonstrate cytokeratin and chromogranin A expression.
EGFR is a member of the EGF-related tyrosine kinase receptor family.19 On ligand binding, the receptors homodimerize or heterodimerize, which activates the intrinsic tyrosine kinase activity of the receptor. This activation results in the activation of broad downstream signaling cascades, including the Ras-Raf-MAP-kinase pathway, the PI3K-Akt pathway, and the STAT pathway. These pathways strongly stimulate cell proliferation, differentiation, survival, angiogenesis, and migration.19,26
Cetuximab, gefitinib, and erlotinib, chemotherapeutic agents that target EGFR, have been administered to patients with cancer (eg, non–small cell lung cancer27). EGFR gene mutations have been reported in patients with non–small cell lung cancer, and the status of these mutations has been correlated with the clinical response to tyrosine kinase inhibitors such as gefitinib.28 Kotoula et al20 reported that mutations of the EGFR locus could be detected in human ACCs, but our study did not detect any mutations in myxoid adrenocortical neoplasms. Only abnormal EGFR protein expression was detected in ACC; the effectiveness of EGFR-targeted therapy for ACC still needs to be confirmed.
EGFR gene amplification and structural genetic alterations have been reported in several adenocarcinoma types, including non–small cell lung cancer, glioblastoma, pancreatic cancer, and squamous cell carcinoma of the head and neck. In our series of cases, an increased EGFR copy number was detected only in a single ACA, which was actively growing with a Ki-67 index of 3% and focal areas of capsular invasion. EGFR amplification was not found in any myxoid adrenocortical neoplasms, suggesting that EGFR amplification may be rare in myxoid adrenocortical neoplasms.
EGFR immunoreactivity was significantly more abundant in myxoid ACCs than in myxoid ACAs, which is consistent with a previous report.21 Because no relationship was found between EGFR protein expression and EGFR mutations or changes in gene copy number, the mechanism underlying the increased EGFR expression remains to be elucidated.
To the best of our knowledge, this is the first study in which the significance of EGFR alterations, including EGFR somatic mutations, gene amplification, and protein expression, in myxoid adrenocortical neoplasms was studied.
Similar to other endocrine neoplasms in other organs, differentiation between benign and malignant adrenocortical tumors is often difficult. Hough et al29 analyzed the presence of histologic and nonhistologic features associated with malignant behavior in adrenocortical tumors. The most significant of these were clinical evidence of weight loss, broad fibrous bands traversing the tumor, a diffuse growth pattern, vascular invasion, tumor cell necrosis, and tumor mass.29 The criteria for malignancy proposed by Weiss30 in 1984 and restudied by Weiss et al31 in 1989 included a combination of the following 9 features: nuclear grade; a mitotic rate greater than 5 per 50 HPFs; atypical mitoses; clear cells comprising no more than 25% of the tumor; a diffuse architecture; microscopic necrosis; and the invasion of venous, sinusoidal, and capsular structures. Page et al32 proposed that the criteria of Hough et al,29 Weiss,30 or Weiss et al31 may be supplanted by new criteria and new technology, such as flow cytometry.
A study by Evans and Vassilopoulou-Sellin,33 which examined 56 adrenocortical neoplasms with a minimum of 5 years of follow-up, found that adenomas typically had a maximal mitotic rate of fewer than 2 mitotic figures per 10 HPFs, which was different from the 5 per 50 HPFs proposed by Weiss30 and Weiss et al31 (1 mitosis per 10 HPFs), a prominent small nest growth pattern, predominantly clear or foamy cytoplasm, and no tumor necrosis. In contrast, carcinomas were characterized by at least 4 mitotic figures (often many more) per 10 HPFs, lack of a nested growth pattern, a dominance of compact eosinophilic cells, and tumor necrosis.33 In addition, Lack34 summarized that important histologic factors in the diagnosis of ACCs were high mitotic count (particularly atypical mitotic figures), vascular invasion, and tumor necrosis. However, it is worth noting that several studies have demonstrated that many pediatric adrenocortical neoplasms with worrisome histologic findings associated with malignancy in adults, such as necrosis, capsular invasion, vascular invasion, increased mitotic rate, and even atypical mitotic figures, ultimately prove to behave in a clinically benign manner.35 So, it is important for pathologists to know that a combination of histologic findings combined with clinical features was most useful in making a diagnosis of ACC. In this study, we observed that a mucinous background was more likely to be observed in ACCs than in ACAs. Overall, we found that all of these findings are applicable to the differential diagnosis of myxoid ACAs and carcinomas.
The differential diagnosis of a myxoid neoplasm in the retroperitoneum includes chordoma, carcinoma with myxoid areas, myxoma, atypical lipomatous tumor with myxoid changes, benign or malignant nerve sheath tumor, myxoid leiomyoma and leiomyosarcoma, extraskeletal myxoid chondrosarcomas, gastrointestinal stromal tumor, and myxoid malignant fibrous histiocytoma.4,36 Careful histopathologic examination of the tumors and better immunohistochemical stains and/or ultrastructural studies can help distinguish these lesions.
Myxoid changes in adrenocortical neoplasms were rare but could be seen in benign and malignant adrenocortical neoplasms. The usual clinical and histologic features could be used to classify the lesions as benign or malignant. Clinicopathologic correlations may be helpful, but special stains and immunohistochemical examinations may be necessary to conclusively establish the diagnosis. To the best of our knowledge, this is the first analysis of EGFR mutations and the EGFR copy number in myxoid adrenocortical neoplasms. In addition, we also tested EGFR protein expression by using immunohistochemical studies. Moreover, this is the largest series of myxoid adrenocortical neoplasm cases in an Asian population in which EGFR alterations have been analyzed. EGFR overexpression was the most frequent EGFR defect in myxoid ACCs; EGFR mutations and changes in the EGFR copy number were minor events.
We thank Yufeng Luo and Shuying Zhang for technical support.
Supported by the National Science and Technology Support Project (the 11th Five-Year Plan) of China (grant 2006BAI02A14).
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}